Significance of Genome Sequencing for African Orphan Crops: the case of Tef
Sonia Plaza, Eligio Bossolini and Zerihun Tadele
Institute of Plant Sciences, University of Bern, Altenbergrain 21, 3013 Bern, Switzerland (Email: sonia.plaza@ips.unibe.ch; eligio.bossolini@ips.unibe.ch; zerihun.tadele@ips.unibe.ch)
Abstract
Knowledge on genome sequence is of paramount importance for the application of modern genetic improvement programs. A genome sequence can provide a better understanding of the tolerance mechanisms conferring adaptability to adverse agronomic conditions. In order to unleash the potential of genetic improvement, we have initiated the Genome Sequencing Project on Tef (Eragrostis tef), an African under-researched crop. Tef is a small grain cereal widely cultivated in the Horn of Africa, particularly in Ethiopia. The crop is tolerant to several biotic and abiotic stresses. Tef performs better than other cereals in poorly drained vertisols, dominant in the highlands of Ethiopia. Despite its importance, tef suffered from a lack of genomic research and is commonly classified as an ‘orphan crop’. The first five runs made using the 454 Next Generation Sequencing Platform generated each about 400Mbp. After assembly the sequences are 244 Mbp with an average size of 610 bp. In order to validate the quality of the sequences, we analysed the phylogenetic relationship between the so called ‘Green Revolution’ gene from major crops and the orthologous tef gene. The results from the pilot sequencing showed promising results and call for the completion of sequencing the whole genome of tef at reasonable cost.
Keywords: Genome sequencing, Next Generation Sequencing (NGS), Eragrostis tef, gibberellic acid insensitive, shotgun sequencing, paired-end sequencing
Introduction
Under-utilized or under-researched crops, commonly referred as ‘orphan crops’ are those crop plants that for historical and economical reasons did not benefit from intensive breeding programs. These plants did not benefit from the genetic improvements due to Green Revolution that dramatically increased cereal production. Genetic improvements have been hindered by a lack of research investments. Molecular tools like genetic maps, mutant collections, molecular markers, DNA libraries, and genomic sequences were so far not affordable. With the advent of the second generation sequencing platforms based on nanotechnologies, the generation of sequence information become cheaper. The number of organisms whose genome has been or is being sequenced is rapidly growing. In this new scenario, also crop plants with a marginal economical importance can profit from these new technologies (see Table 1). At least two factors influence the choice of species being sequenced: i) genome size: due to technical limitations in most cases organisms with smaller genome size are chosen for sequencing, and ii) genetic distance: the phylogenetic distance from previously sequenced organisms is also important for ease of transfer of information on genome structure and function from a well-characterized species to the related but less studied species.
Importance of tef in the economy of developing country
Tef is a staple food in East Africa particularly in Ethiopia where it is annually cultivated on about 2.5 million hectares of land [11]. The tef plant possesses a number of advantages: i) the crop performs better than other major cereals under extreme environmental conditions such as drought and water-logging; ii) the tef flour does not contain gluten, making it particularly suitable in the diet of celiac consumers; iii) the seeds are less attacked by storage pests, hence, can be stored for long time without losing viability; and iv) the crop requires small agronomical inputs, making it particularly suitable for extensive and sustainable agriculture [12]. Tef is therefore important in maintaining food security in rapidly evolving environmental conditions, by allowing its cultivation in adverse regions where high input crops would normally fail.
Nevertheless, tef presents a major noteworthy disadvantage: its yield is very low. In Ethiopia, where tef is extensively grown, the national average yield is only 0.9 t/ha as compared to 1.7 t/ha for wheat [13]. The major yield limiting factor is in tef lodging, i.e. the permanent displacement of the stem from the upright position. Tef has a tall and weak stem that falls on the ground due to wind and rain. The application of nitrogen fertilizers also aggravates lodging. The lodged plants produce inferior yield both in terms of quantity and quality. Because of its very local cultivation and marginal economical relevance, Green Revolution was not implemented on this particular crop. The majority of research on tef has been so far undertaken in Ethiopia, using mainly conventional breeding techniques. Up to date, the Ethiopian research institutes released 17 improved cultivars to farmers, seven of which are from hybridization while the remaining ten from selection [14].
Need for sequencing the tef genome
Whole genome sequencing is particularly important for the improvement of the orphan crops such as tef for which few genetic and genomic resources are available. Tef is an allotetraploid species (2n = 4x = 40). To date the two diploid ancestors that originated its genome by hybridization remain uncertain [15]. Despite its ploidy level, tef maintains a medium genome size (730Mbp) [16], almost the same size as sorghum [10]. However, tef is allotetraploid whereas sorghum is diploid, suggesting a double gene density in tef. Phylogenetically, sorghum is also the closest sequenced genome to tef and represents a template for comparative studies. Partial transcriptome sequence information, consisting of an EST collection of 3603 sequences has been developed and used to construct a genetic map derived from an interspecific cross (Eragrostis tef x E. pilosa) [17,18]. From this work, a QTL analysis on 22 yield-related and morphological traits has been performed [19]. Given the available molecular tools and the new cost-effective sequencing technologies, sequencing the gene-dense genome of tef has become not only feasible but also a compelling necessity. Some of the specific advantages of sequencing the tef genome are i) sequence information for any gene of interest will be available, including promoter and terminator regions, hence avoiding the need of predicting gene sequences by homology to well-characterized model plants like rice and sorghum; ii) being tef a tetraploid, the complete genome sequence will facilitate the isolation of a specific gene, while distinguishing between the two possible orthologous copies. This is of special interest during the implementation of TILLING (Targeting Induced Local Lesions In Genomes) techniques [20], where the presence of an orthologous variant could be erroneously interpreted as a mutant; iii) genomic sequence information will provide a template for the development of genetic markers such as Single Nucleotide Polymorphisms (SNPs) and Simple Sequence Repeats (SSRs). These high-throughput molecular markers are more and more employed for marker-assisted breeding, for the construction of high density genetic maps and for linkage disequilibrium studies on diverse germplasm; molecular applications such as gene cloning, gene over-expression, or down-regulation of genes of choice will be achieved with minimum effort and finally; iv) tef is tolerant to many adverse climatic and soils conditions, especially to water-logging. The understanding of the molecular basis of tolerance mechanisms gained from the genome sequence of this unique plant can possibly be transferred to other economically important crops with higher input requirements.
The Tef Genome Sequencing Initiative
The Tef Genome Sequencing initiative began at the end of 2009. The early maturing and wide adaptable tef cultivar DZ-Cr-37 was selected for the sequencing. The main objectives of the tef sequencing initiative are, i) to sequence, analyze and annotate the tef genome, and ii) to make publicly available all generated information. The strategies to be used in sequencing the tef genome are the following: i) fragment sequencing up to 10x coverage of the genome; ii) paired-end sequencing of libraries with different insert sizes. This strategy will help to overcome assembly problems related to polyploidy; iii) BAC sequencing will be done to improve the final assembly if necessary.
The Pilot Sequencing Project
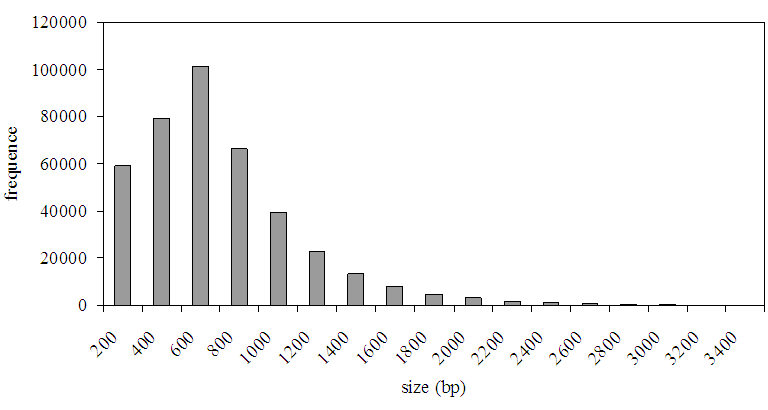
In order to investigate the quality of the sequencing obtained from the Next Generation Sequencing (NGS), we run a pilot project using 5 runs of the Roche 454 GS-FLX Platform [21] producing a total of 245 Mbp reads. These were clustered and assembled using the Newbler software from Roche, with the default settings of 98% similarity and 50 bp of overlap. The assembly consists of 402 304 contigs with a minimum length 100 bp. Of these, 214 750 contigs were longer than 500 bp, the biggest contig has currently a length of 17 200 bp (see Fig. 1 for the sequence length distribution of the assembly).
A blastx search [22] with a threshold of e-10 was performed on tef contigs to identify sequences with homology to the proteome of sorghum (version sbi.1.4, 34496 loci [10]). The low stringency threshold of e-10 was adopted to compensate for partial and fragmented tef sequences. Under these blast parameters, 92% of the sorghum loci had a hit in our tef contigs with an e-10. Nevertheless, the low stringency threshold used can lead to misidentification of paralogous and orthologous gene copies [23]. To exclude multiple hits to non-orthologous gene family members, we repeated the blast search using a subset of 5796 sorghum genes with low homology to any other gene in the genome. This set consists of only those genes that do not have any other match in the sorghum genome with a blastn threshold of e-10. Within this ‘non-redundant’ subset only 55% of the genes had a hit in the tef database.
Due to our interest to obtain semi-dwarf tef lines that are tolerant to lodging, we used sequences from the pilot sequencing to analyze the so called Green Revolution genes that boosted the yield of wheat and rice in the 1960s and 70s. As case study we focused on the Gibberellic Acid Insensitive (GAI, [24]) gene from the model plant Arabidopsis. The corresponding genes in crops species are Reduced height (Rht) in wheat, Dwarf 8 (d8) in maize, and Slender 1 (SLR1) in rice. The mutants of this particular gene in the indicated species are affected in size and do not respond to the exogenous application of gibberellic acid ([25 to 29]). In order to validate the quality of the sequencing, the orthologous copy of the GAI gene was identified from the tef contigs. Tef amplicons of genomic and coding DNA of the GAI gene were obtained with specific primers (EtGAI_S1: 5’- ATGGAAGCGCGAGTACCAAG -3’ and EtGAI_AS1: 5’- ACCACCGGTAAGGAGATCG -3’) designed on the contigs generated within this pilot sequencing project. Sequences of the orthologous gene from related grasses were obtained from the NCBI database [30] (sorghum: “XM_002466549”, sugarcane: “DQ062091”, maize: “AJ242530”, pearl millet: “FJ011693.1” and rice: “AB262980”). The alignment of these sequences with the contigs of tef was done using ClustalW [31]. Then it was manually refined and pairwise comparison were confirmed with BLAST [22]. A genetic tree was generated with the program MEGA 4.1 [32], using the neighbour joining method under Jukes-Cantor model with a bootstrap value of 1000 replications.
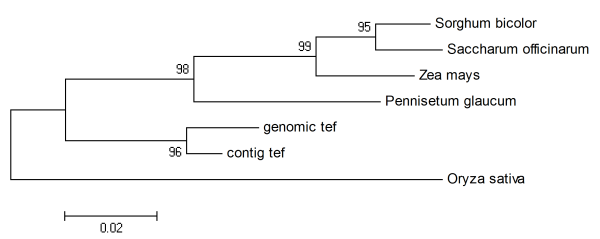
Fig 2 shows the current state of work for GAI homology in tef. From the sequences made so far, two tef contigs with the size of 730 and 398 bp having homology to the 5’ and 3’ regions of the GAI gene, respectively, were obtained. By using primers designed from the first contig, we amplified a single amplicon (490bp) showing a high homology with the tef contig (99%) proving the usefulness of using the contigs to design specific primers. The small differences observed between the sequences of the amplicon and the contig could be explained by either having amplified the other allelic copy of GAI homolog gene as tef is an allotetraploid species or might be due to errors present in the sequencing or in the assembly. Moreover, a similar phylogenetic tree [33] was obtained for the region of GAI homolog genes corresponding to the amplified sequence of tef (Fig. 3 and 4, for the homology percent and the phylogenic tree, respectively). No sequence of this gene was available in the genbank database [30] for finger millet (Eleusine coracana), a closer species to tef. The cluster comprising the two sequences of tef (the contig and amplicon) were closest to pearl millet, followed by a cluster formed by maize, sorghum and sugarcane.
The prospects of genome sequencing on orphan crops
Recent progress in the development of genome-scale data set for several crop species offer important new possibilities for crop improvement. This will enable breeders and biotechnologists to more rapidly and precisely target genes that underlie key agronomic traits, and with such knowledge to develop molecular assays that are both relevant and of appropriate scale for breeding application. The main target will be abiotic and biotic stresses limiting crop productivity [34].
In the present study, the identification and PCR amplification of a gene using primers designed on a contig has shown the utility of the sequence information that has so far been generated, although the status of the assembly is still largely fragmented and incomplete. The unfinished status of the genome sequence resulted in a number of limitations including, i) most genes are partial and in many cases difficult to identify the orthologous copies of each tef sub-genome as tef is an allotetraploid; ii) the identification of gene family members is hindered by the fragmented nature of the current assembly and; iii) several genes present in sorghum and rice could not be identified in tef. Given the available data, it is not possible to determine whether these genes are indeed not present in tef or they have not been sequenced yet.
In conclusion, we have described the first genome sequence initiative on the under-studied African crop tef. This is of paramount importance for the genetic improvement of this staple crop. As tef is tolerant to abiotic and biotic stresses, the information from the sequencing will shed light on the molecular mechanisms conferring tolerance to a variety of stresses. This information may be transferred to other crops that are less adapted to adverse growing conditions. More investments have to be devolved to the genetic improvement of African neglected crops. Marginal environmental conditions expose African low input agricultural systems to a higher risk than high input modern agriculture. African enduring crops deserve more investments to stabilize future food production especially in the current scenario of climate change.
Acknowledgments
We are very grateful to Syngenta Foundation for Sustainable Agriculture and to the University of Bern for supporting our research on orphan crop tef. The sequencing has been done at the Functional Genomic Center of Zürich.
References
[1] Han, Y. and Korban, S.S. (2008). An overview of the apple genome through BAC end sequence analysis. Plant Molecular Biology 67 (6): 581-588.
[2] Bell, C.J., Dixon, R.A., Farmer, A.D., Flores, R, Inman, J., Gonzales, R.A., Harrison, M.J., Paiva, N.L., Scott, A.D., Weller, J.W. and May, G.D. (2001). The Medicago Genome Initiative: a model legume database. Nucleic Acids Res. 29(1): 114-117.
[3] Huang, S., Li, R., Zhang, Z., Li, L., Gu, X., Fan, W., Lucas, W.J., Wang, X., Xie, B., Ni, P., Ren, Y., Zhu, H., Li, J., Lin, K., Jin, W., Fei, Z., Li, G., Staub, J., Kilian, A., van der Vossen, E.A.G., Wu, Y., Guo, J., He, J., Jia, Z., Ren, Y., Tian, G., Lu, Y., Ruan, J., Qian, W., Wang, M., Huang, H., Li, B., Xuan, Z., Cao, J., Wu, A.Z, Zhang, J., Cai, Q., Bai,Y., Zhao, B., Han, Y., Li, Y., Li, X., Wang, S., Shi, Q., Liu, S., Cho, W.K., Kim,J.-Y., Xu, Y., Heller-Uszynska, K., Miao, H., Cheng, Z., Zhang, S., Wu, J., Yang, Y., Kang, H., Li, M., Liang, H., Ren, X., Shi, Z., Wen, M., Jian, M., Yang, H., Zhang, G., Yang, Z., Chen, R., Liu, S., Li, J., Ma, L., Liu, H., Zhou, Y., Zhao, J., Fang, X., Li, G., Fang, L., Li, Y., Liu, D., Zheng, H., Zhang, Y., Qin, N., Li, Z., Yang, G., Yang, S., Bolund, L., Kristiansen, K., Zheng, H., Li, S., Zhang, X., Yang, H., Wang, J., Sun, R., Zhang, B., Jiang, S., Wang, J., Du, Y. and Songgang Li, S. (2009). The genome of the cucumber, Cucumis sativus L. Nature Genetics, 41: 1275-1281.
[4] Palmer, L.E., Rabinowicz, P.D., O’Shaughnessy, A.L., Balija, V.S., Nascimento, L.U., de la Bastide, M., Martienssen, R.A. and McCombie, W.R. (2003). Maize genome sequencing by methylation filtration. Science, 302:2115-2117.
[5] Ming, R., Hou, S., Feng, Y., Yu, Q., Dionne-Laporte, A., Saw, J.H., Senin, P., Wang, W. and Ly, B.V. (2008). The draft genome of the transgenic tropical fruit tree papaya (Carica papaya Linnaeus) Nature, 452: 991-996.
[6] Tuskan, G.A., DiFazio, S., Jansson, S., Bohlmann, J., Grigoriev, I., Hellsten, U., Putnam, N., Ralph, S., Rombauts, S., Salamov, A., Schein, J., Sterck, L., Aerts, A., Bhalerao, R.R:, Bhalerao, R.P., Blaudez, D., Boerjan, W., Brun, A., Brunner, A., Busov, V., Campbell, M., Carlson, J., Chalot, M., Chapman, J., Chen, G.-L., Cooper, D., Coutinho, P.M., Couturier, J., Covert, S., Cronk, Q., Cunningham, R., Davis, J., Degroeve, S., Déjardin, A., DePamphilis, C., Detter, J., Dirks, B., Dubchak, I., Duplessis, S., Ehlting, J., Ellis, B., Gendler, K., Goodstein, D., Gribskov, M., Grimwood, J., Groover, A., Gunter, L., Hamberger, B., Heinze, B., Helariutta, Y., Henrissat, B., Holligan, D., Holt, R., Huang, W., Islam-Faridi, N., Jones, S., Jones-Rhoades, M., Jorgensen, R., Joshi, C., Kangasjärvi, J., Karlsson, J., Kellenher, C., Kirkpatrick, R., Kirst, M., Kohler, A., Kalluri, U., Larimer, F., Leebens-Mack, J., Leplé, J.-C., Locascio, P., Lou, Y., Lucas, S., Martin, F., Montanini, B., Napoli, C., Nelson, D.R., Nelson, C., Nieminen, K., Nilsson, O., Pereda, V., Peter, G., Philippe, R., Pilate, G., Poliakov, A., Razumovskaya, J., Richardson, P., Rinaldi, C., Ritland, K., Rouzé, P., Ryaboy, D., Schmutz, J., Schrader, J., Segerman, B., Shin, H., Siddiqui, A., Sterky, F., Terry, A., Tsai, C.-J., Uberbacher, E., Unneberg, P., Vahala, J., Wall, K., Wessler, S., Yang, G., Yin, T., Douglas, C., Marra, M., Sandberg, G., Van de Peer, Y. and Rokhsar, D. (2006). The genome of black cottonwood, Populus trichocarpa (Torr. & Gray). Science, 313: 1596-1604.
[7] Yuan, Q., Ouyang, S., Wang, A., Zhu, W., Maiti, R., Lin, H., Hamilton, J., Haas, B., Sultana, R., Cheung, F., Wortman, J. and Buell, C.R. (2005). The Institute for Genomic Research Osa1 Rice Genome Annotation Database. Plant Physiology, 138: 18-26.
[8] Ouyang, S. Zhu, W., Hamilton, J., Lin, H., Campbell, M., Childs, K., Thibaud-Nissen, F., Malek, R.L., Lee, Y., Zheng, L., Orvis, J., Haas, B., Wortman, J. and Buell, C.R. (2007). The TIGR Rice Genome Annotation Resource: improvements and new features. Nucleic Acids Research, 35:D883-D887.
[9] Bedell, J.A., Budiman, M.A., Nunberg, A., Citek, R.W., Robbins, D., Jones, J., Flick, E., Rohlfing, T., Fries, J. Bradford, K., McMenamy, J., Smith, M., Holeman, H., Roe, B.A., Wiley, G., Korf, I.F., Rabinowicz, P.D., Lakey, N., McCombie, W.R., Jeddeloh, J.A., Martienssen, R.A. (2005). Sorghum genome sequencing by methylation filtration. PLOS, 3: 103-115.
[10] Paterson A.H., Bowers, J.E., Bruggmann, R., Dubchak, I., Grimwood, J., Gundlach, H., Haberer, G., Hellsten. U., Mitros, T., Poliakov, A., Schmutz, J., Spannagl, M., Tang, H., Wang, X., Wicker, T., Bharti, A.K., Chapman, J., Feltus, A.F., Gowik, U., Grigoriev, I.V., Lyons, E., Maher, C.A., Martis, M., Narechania, A., Otillar, R.P., Penning, B.W., Salamov, A.A., Wang, Y., Zhang, L., Carpita, N.C., Freeling, M., Gingle, A.R., Hash, C.T., Keller, B., Klein, P., Kresovich, S., McCann, M.C., Ming, R., Peterson, D.G., Rahman, M., Ware, D., Westhoff, P., Mayer, K.F.X., Joachim Messing, J. and Rokhsar, D.S. (2009). The Sorghum bicolor genome and the diversification of grasses. Nature, 457: 551-556.
[11] FAO. (2009). Special Report. FAO/WFP Crop and food security assessment mission to Ethiopia. 21. Januar 2009. http://www.fao.org/docrep/011/ai477e/ai477e00.htm
[12] Habtegebrial, K. and Singh, B.R. (2006). Effects of timing of nitrogen and sulphur fertilizers on yield, nitrogen, and sulphur contents on Tef (Eragrostis tef (Zucc.) Trotter). Nutr. Cycl. Agroecosyst., 75: 213-222.
[13] Feyissa, R. (1999). Community seed banks and seed exchange in Ethiopia: a farmer-led approach. In: Participatory approaches to the conservation and use of plant genetic resources. Friis-Hansen and Sthapit (eds) pp: 142-148.
[14] Assefa, K.; Belay, G.; Tefera, H.; Yu, J.-K. and Sorrells, M.E. (2009). Breeding tef: conventional and molecular approaches. In: new approaches to Plant Breeding of Orphan Crops in Africa: Proceedings of an International Conference, 19-21 September 2007, Bern, Switzerland, Tadele Z (ed.) pp: 21-41.
[15] Ingram, A.L. and Doyle, J.J. (2003). The origin and evolution of Eragrostis tef (Poaceae) and related polyploids: evidence from nuclear waxy and plastic rps16. American Journal of Botany, 90: 116-122.
[16] Ayele, M., Dolezel, J., van Duren, M., Brunnen, H. and Zapata-Arias, F.J. (1996). Flow cytometric analysis of nuclear genome of the Ethiopian cereal tef [Eragrostis tef (Zucc.) Trotter]. Genetics, 98: 211-215. Genome, 49: 365-372.
[17] Yu, J.-K., Sun, Q., La Rota, M., Edwards, H., Tefera, H. and Sorrells, M.E. (2006). Expressed sequence tag analysis in tef [Eragrostis tef (Zucc) Trotter]. Genome, 49: 365-372.
[18] Yu, J.-K., Kantety, R.V., Graznak, E., Benscher, D., Tefera, H. and Sorrells, M.E. (2006). A genetic linkage map for tef [Eragrostis tef (Zucc) Trotter]. Theor Appl Genet, 113: 1093-1102.
[19] Yu, J.-K., Graznak, E., Breseghello, F., Tefera, H. and Sorrells, M.E. (2007). QTL mapping of agronomic traits in tef [Eragrostis tef (Zucc) Trotter]. BMC Plant Biology, 7: 30-43.
[20] Esfeld, K. and Tadele, Z. (this issue). The improvement of African orphan crops through TILLING.
[21] Margulies, M., Egholm,M., Altman W.E., Attiya S., Bader, J.S., Bemben, L.A., Berka, J., Braverman, M.S., Chen, Y.J., Chen, Z., Dewell, S.B., Du, L., Fierro, J.M., Gomes, X.V., Godwin, B.C., He, W., Helgesen, S., Ho, C.H., Irzyk, G.P., Jando, S.C., Alenquer, M.L.I., Jarvie, T.P., Jirage, K.B., Kim, J.B., Knight, J.R., Lanza, J.R., Leamon, J.H., Lefkowitz, S.M., Lei, M., Li, J., Lohman, K.L., Lu, H., Makhijani, V.B., McDade, K.E., McKenna, M.P., Myers, E.W., Nickerson, E., Nobile, J.R., Plant, R., Puc, B.P., Ronan, M.T., Roth, G.T., Sarkis, G.J., Simons, J.F., Simpson, J.W., Srinivasan, M., Tartaro, K.R., Alexander Tomasz, A., Vogt, K.A., Volkmer, G.A., Wang, S.H., Wang, Y., Weiner, M.P., Yu, P., Begley, R.F. and Rothberg, J.M. (2005). Genome sequencing in microfabricated high-density picolitre reactors. Nature, 437: 376–380.
[22] Altschul, S.F., Gish, W., Miller, W., Myers, E.W. and Lipman, D.J. (1990). Basic local alignment search tool. J. Mol. Biol., 215: 403-410.
[23] Salse, J., Abrouk, M., Murat, F., Quraishi, U.M. and Feuillet, C. (2009). Improved criteria and comparative genomics tool provide new insights into grass paleogenomics, Briefings in Bioinformatics, 10: 619-630.
[24] Koorneef, M., Elgersma, A., Hanhart, C.J., van Loenen-Martinet, E.P., van Rign, L. and Zeevaart, J.A.D. (1985). A gibberellin insensitive mutant of Arabidopsis thaliana. Physiol. Plant. 65: 33-39.
[25] Harberd, N.P. and Freeling, M. 1989. Genetics of dominant gibberellin-insensitive dwarfism in maize. Genetics, 121: 827-838.
[26] Winkler, R.G. and Freeling, M. (1994). Physiological genetics of the dominant gibberellin-nonresponsive maize dwarf, Dwarf8 and Dwarf9. Planta, 193: 341-348.
[27] Peng, J., Carol, P., Richards, D.E., King, K.E., Cowling, R.J., Murphy, G.P. and Harberd, N.P. (1997). The Arabidopsis GAI gene defines a signaling pathway that negatively regulates gibberellin responses. Genes Dev., 11: 3194-3205.
[28] Peng, J., Richards, D.E., Hartley, N.M., Murphy, G.P., Devos, K.M., Flintham, J.E., Beales, J., Fish, L.J., Worland, A.J., Pelica, F., Sudhakar, D., Christou, P., Snape, J.W., Gale, M.D. and Harberd, N.P. (1999). “Green revolution” genes encode mutant gibberellin response modulators. Nature, 400: 256-261.
[29] Itoh, H., Ueguchi-Tanka, M., Sato, Y., Ashikari, M. and Matsuoka, M. (2002). The gibberellin signaling pathway is regulated by the appearance and disappearance of SLENDER RICE1 in nuclei. The Plant Cell, 14: 57-70.
[30] www.ncbi.nlm.nih.gov
[31] Larkin, M.A., Blackshields, G., Brown, N.P., Chenna, R., McGettigan, P.A., McWilliam, H, Valentin, F., Wallace, I.M., Wilm, A., Lopez, R., Thompson, J.D., Gibson, T.J. and Higgins, D.G. (2007). ClustalW and ClustalX version 2. Bioinformatics, 23: 2947-2948.
[32] Tamura, K., Dudley, J., Nei, M. and Kumar, S. (2007). MEGA4: Molecular evolutionary genetics analysis (MEGA) software version 4.0. Molecular Biology and Evolution, 24: 1596-1599.
[33] Devos, K.M. (2005). Updating the “Crop Circle”. Current Opinion in Plant Biology, 8: 155-162.
[34] Varshney, R.K., Close, T.J., Singh, N.K., Hoisington, D.A. and Cook, D.R. (2009). Orphan legume crops entre the genomics era! Current Opinion in Plant Biology, 12: 202-210.
Table 1. Genome size and the status of genome sequencing for crop plants
Common name | Scientific name | Polyploidy level | Genome size | Current state of work |
Apple | Malus domestica | 2n=2x=34 | 750 Mb | Genome published [1] |
Barrel medic | Medicago truncatula | 2n=2x=16 | 550 Mb | Genome published [2] |
Canola | Brassica napus | 2n=2x=38 | 1129Mb | Completed |
Cassava | Manihot esculenta | 2n=2x=36 | 760 Mb | Completed |
Cotton | Gossypium raimondii | 2n=2x=26 | 880 Mb | In progress |
Cucumber | Cucumus sativis | 2n=2x=14 | 367 Mb | Genome published [3] |
Grape | Vitis vinifera | 2n=2x=38 | 500 Mb | In progress |
Maize | Zea mays | 2n=2x=20 | 2600 Mb | Genome published [4] |
Papaya | Carica papya | 2n=2x=18 | 372 Mb | Genome published [5] |
Peach | Prunus persica | 2n=2x=16 | 270 Mb | Incomplete |
Poplar | Populus trichocarpa | 2n=2x=38 | 480 Mb | Genome published [6] |
Potato | Solanum tuberosum | 2n=4x=48 | 840 Mb | Completed |
Rice | Oryza sativa | 2n=2x=24 | 430 Mb | Genome published [7,8] |
Sorghum | Sorghum bicolor | 2n=2x=20 | 736 Mb | Genome published [9,10] |
Soybean | Glycine max | 2n=2x=40 | 1115 Mb | Completed |
Tomato | Solanum lycopersicum | 2n=2x=24 | 950 Mb | Completed |
Fig 1. Frequency distribution of the tef contigs after assembly
Fig 2. Alignment of tef contigs to genomic sorghum sequence. The numbers correspond to the genomic position in sorghum. The numbers in the boxes correspond to the two contigs.
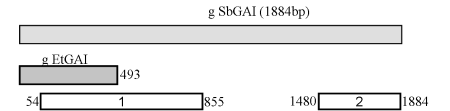
Fig 3. Percent of homology between the two tef contigs and homologs of GAI gene of sorghum, maize, sugarcane, pearl millet and rice. Accessions of full length sequences (except for pearl millet) were obtained from NCBI and indicated in the text.
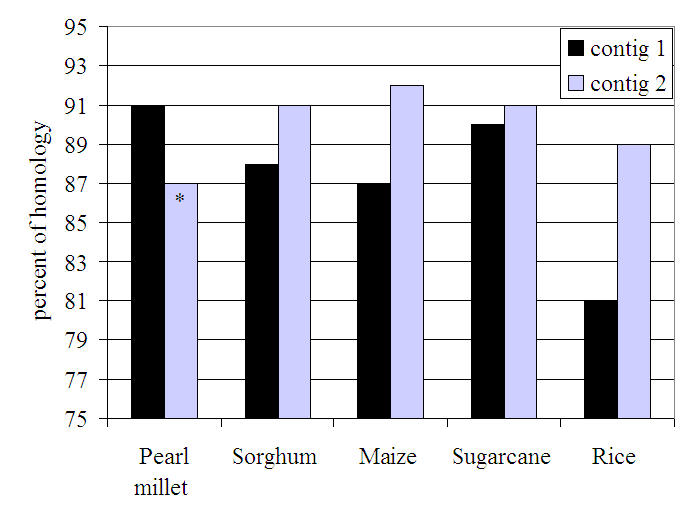
* As partial sequence, only 250bp were compared.
Fig 4. Phylogenic analysis of GAI genes. Numbers above the branches are bootstrap values.